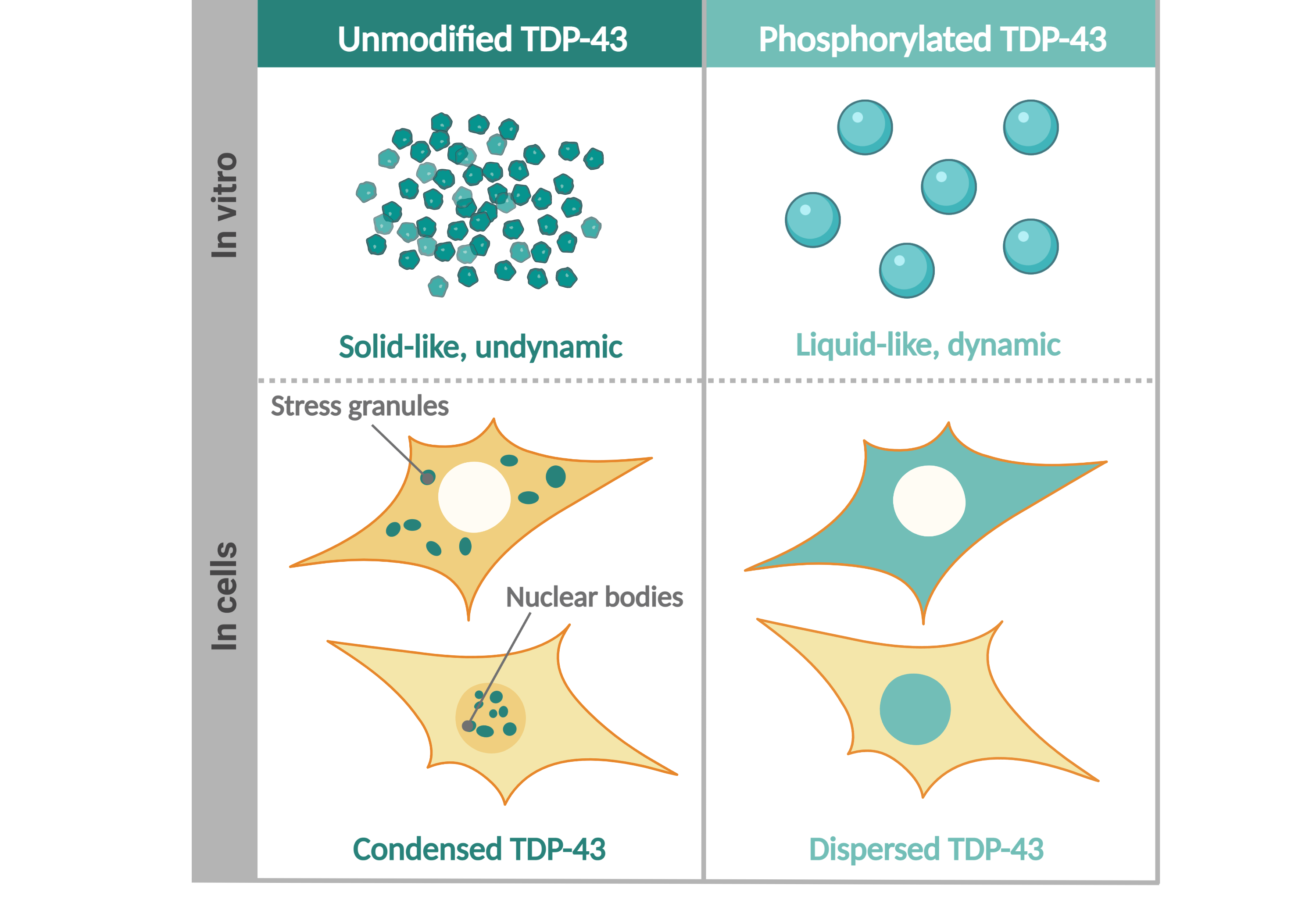
Figure: C-terminal hyperphosphorylation of TDP-42, a pathological hallmark of several neurodegenerative disorders, enhances the liquidity of TDP-43 condensates and suppresses TDP-43 condensation and aggregation, shedding a new light on this disease-linked post-translational modification.
Ref: Gruijs da Silva LA, Simonetti F, Hutten S, Riemenschneider H, Sternburg EL, Pietrek LM, Gebel J, Doetsch V, Edbauer D, Hummer G, Stelzl LS, Dormann D. Disease-linked TDP-43 hyperphosphorylation suppresses TDP-43 condensation and aggregation. EMBO J, in press
read on bioarxiv